Sjukdom/skada/diagnos
PMM2-CDG (tidigare CDG-Ia) är den vanligaste sjukdomen i en grupp biokemiska ämnesomsättningssjukdomar som först beskrevs på 1980-talet, gemensamt kallade CDG-syndromen. CDG är en förkortning av det engelska namnet ’congenital disorders of glycosylation’, som betyder medfödda glykosyleringsstörningar.
För närvarande (2014) finns ett drygt 100-tal olika former av CDG-syndromen beskrivna. PMM2-CDG ingår i en grupp av CDG-syndromen, kallad N-glykosyleringsstörningar, som idag omfattar cirka 50 olika former.
Sjukdomen ger symtom från flera olika organsystem som centrala nervsystemet, muskulaturen, levern, mag-tarmkanalen, hormonsystemen och ögonen. Under nyföddhetsperioden och barnaåren kan PMM2-CDG vara livshotande, medan sjukdomsbilden hos vuxna är ganska stabil med lite försämring.
Förekomst
PMM2-CDG beräknas finnas hos knappt två barn per 100 000 nyfödda, vilket skulle innebära att det föds i genomsnitt 1 – 2 barn med sjukdomen varje år i Sverige. Man känner till ett 25-tal personer med PMM2-CDG i landet. De övriga formerna av CDG-syndromen är ännu ovanligare och har hittills bara diagnostiserats hos ett fåtal barn i Sverige.
Orsak till sjukdomen/skadan
De olika CDG-syndromen beror på fel i en process som kallas glykosylering. Glykosylering innebär att vissa proteiner och fetter (lipider) blir bärare av komplexa sockerarter (glykoproteiner respektive glykolipider). Glykoproteinerna består av en proteindel på vilken sockermolekyler som sticker ut som små antenner är fästade. Störningen i glykoliseringsprocessen gör att många viktiga glykoproteiner och glykolipider i kroppen blir felaktigt uppbyggda och delvis saknar sina sockerantenner. Dessa biokemiska fel påverkar ofta kraftigt utvecklingen av nervsystemet, men även andra organ påverkas också för det mesta, till exempel levern, skelettet, musklerna, sköldkörteln, hjärtat och bukspottkörteln.
PMM2-CDG orsakas av en förändring (mutation) i genen PMM2 (16p13.2), som är en mall för tillverkningen av (kodar för) enzymet fosfomannomutas 2 (PMM2). Enzymer är proteiner som reglerar hastigheten av olika kemiska reaktioner i kroppen utan att själva förbrukas. Fosfomannomutas 2 omvandlar sockermolekylen mannos-6-fosfat till mannos-1-fosfat, vilket är nödvändigt för att mannos ska kunna användas vid bildandet av komplexa sockerkedjor (oligosackarider). Resultatet blir att en del av kroppens glykoproteiner får allt för få sockerkedjor bundna till sig.
Typen av kolhydratdefekt skiljer sig åt mellan de olika formerna av N-glykosyleringsstörning. Numera delas sjukdomsformerna in i två grupper, typ I och typ II, beroende på om felet uppstått före eller efter att sockermolekylen bundits till proteinet. Typ I och II delas i sin tur in i olika undergrupper.
Fler än hundra olika mutationer är beskrivna i PMM2, varav de flesta är punktmutationer som leder till ett PMM2-enzym med försämrad funktion (missense mutations). Den vanligaste mutationen är R141H (aminosyran arginin på plats 141 i PMM2 har bytts ut till en histidin). Den leder till att enzymaktiviteten helt saknas (nollmutation). Att ha två gener med denna mutation är inte förenligt med överlevnad.
Ärftlighet
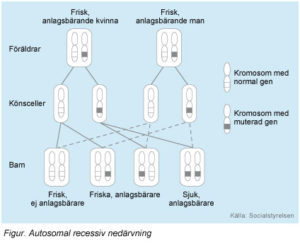
PMM2-CDG och nästan alla andra CDG-syndrom nedärvs autosomalt recessivt, vilket innebär att båda föräldrarna är friska bärare av en förändrad (muterad) gen. Vid varje graviditet med samma föräldrar är sannolikheten 25 procent att barnet får den muterade genen i dubbel uppsättning (en från varje förälder). Barnet får då sjukdomen. Sannolikheten att barnet får den muterade genen i enkel uppsättning är 50 procent. Då blir barnet, liksom föräldrarna, frisk bärare av den muterade genen. Sannolikheten att barnet varken får sjukdomen eller blir bärare av den muterade genen är 25 procent.
Vid studier av PMM2-CDG har det visat sig att 1/3 av fostren till föräldrar som är bärare av PMM2-mutationer får sjukdomen. I ovanliga fall har en av mutationerna uppstått i en könscell (germline mutation) och risken för återupprepning är då i princip ingen. Det är därför viktigt att även föräldrarna erbjuds genetisk utredning i samband med att PMM2-CDG diagnosticeras (se under rubriken Diagnostik).
Symtom
Störningen i ämnesomsättningen leder till skador i storhjärnan (cerebral atrofi), lillhjärnan och hjärnstammen (olivopontocerebellär atrofi), perifera nerverna, näthinnan, skelettet, levern, könskörtlarna och ofta är även hjärtat, njurarna, bukspottkörteln och sköldkörteln påverkade. Symtomen är mycket varierande i olika åldrar, liksom mellan olika personer med sjukdomen. Cirka 20 procent av barnen som föds med PMM2-CDG avlider under de första fem levnadsåren i någon komplikation, som blödning eller organsvikt. Någon exakt koppling mellan vilken mutation man har och hur sjuk man blir finns inte, men mutationer som ger ett PMM2-enzym med relativt hög kvarvarande aktivitet ger oftast ett lindrigare sjukdomsförlopp.
Vid PMM2-CDG finns en typisk form med likartad symtombild (klassisk PMM2-CDG). Under de senaste 10 åren har det dock kommit publikationer som beskriver barn med en mycket varierande symtombild. I Sverige dominerar den klassiska formen, i vilken symtomen brukar delas in i fyra olika stadier:
De första levnadsåren
De nyfödda barnen har karaktäristiska fettkuddar på stjärten och hårt och gropigt underhudsfett, som ger huden ett apelsinskalsliknande utseende (så kallat peau d’orange). Ett annat typiskt tecken är inverterade bröstvårtor. Skelning är också vanligt och beror på svaghet i yttre ögonmusklerna. Många barn har under de första levnadsmånaderna, innan de kan fixera blicken, rullande och irrande ögonrörelser. Muskulaturen är ofta slapp och rörligheten i lederna nedsatt. Grovmotoriken är sent utvecklad och barnen har svårt att rulla runt, sitta och krypa.
Barn med syndromet är ofta ospecifikt sjuka och har svårt att öka i vikt och växa. De har dålig aptit, kräks och får diarré, vilket kan bero på att levern är påverkad, eftersom nästan alla har nedsatt leverfunktion. Leverpåverkan kan i sin tur utvecklas till fett- eller skrumplever. Ibland är låg blodsockerhalt (hypoglykemi) det första tecknet på sjukdomen. Det kan yttra sig som skakighet, irritabilitet och kramper. Ungefär hälften av barnen får vätska i hjärtsäcken, vilket kan leda till hjärtsvikt. Störningar i blodets förmåga att koagulera kan ge blödningar eller blodproppar. De första åren kan sjukdomen vara mycket allvarlig och familjesituationen omtumlande, särskilt om barnet saknar diagnos.
Barndomen
Den kognitiva och motoriska utvecklingen är starkt försenad, och en måttlig till svår utvecklingsstörning förekommer hos de flesta med syndromet. Barnen beskrivs ofta som glada och sociala, men uttalsvårigheter (dysartri) kan göra att de får problem att göra sig förstådda.
Balans- och koordinationssvårigheter (ataxi) beror på att lillhjärnan förtvinar (lillhjärnsatrofi) och på perifer nervskada (neuropati), som ger svaghet i benmuskulaturen. Få barn lär sig gå själva, men många kan förflytta sig av egen kraft genom att hasa eller krypa. De flesta blir duktiga på att använda manuell eller elektrisk rullstol.
Organsvikten avtar successivt, men hälften av barnen utvecklar epilepsi och vart tredje barn drabbas av strokeliknande anfall. De får då, ofta i samband med någon infektion, plötsliga attacker med medvetslöshet eller kraftig påverkan på medvetandet. Även om barnen får neurologiska symtom, som halvsidesförlamning eller blindhet, återhämtar de sig oftast efter några timmar eller dagar.
Störningarna i blodets levringsförmåga kvarstår dock.
Pigmentförändringar i näthinnan (retinitis pigmentosa) tillkommer och ger inskränkt synfält och försämrat mörkerseende. Många av barnen har också brytningsfel.
Avvikelser i skelettet, främst i rygg och bröstkorg (kyfos och skolios), utvecklas gradvis. Till syndromet hör också speciella ansiktsdrag, med stora öron, tunn överläpp och käkar som efter hand blir mer framträdande.
Tonåren
De medicinska komplikationerna minskar under tonåren, men epilepsi kan förekomma, och svagheten i benen blir mer uttalad. Den beror på en försämrad nervfunktion men också på att kroppsvikten ökar och ställer krav på mer benstyrka, samtidigt som musklerna i benen är tunna och dåligt utvecklade.
Flickor med syndromet kommer inte i puberteten (hypergonadotrop hypogonadism), eftersom äggstockarna tillbakabildas tidigt i livet. Pojkarna har en normal men ofta försenad pubertet och små testiklar. Könskörtlarnas nedsatta funktion ger brist på könshormoner, som i sin tur gör att skelettet hos de flesta visar tecken på skörhet eller urkalkning och att ryggen och bröstkorgen får ett hoppressat utseende. Personer med syndromet blir därför uttalat kortväxta, och få kan gå utan hjälpmedel.
Vuxenlivet
Vuxna med PMM2-CDG har ett varaktigt, stabilt tillstånd. De kognitiva funktionsnedsättningarna försämras inte, inte heller talsvårigheterna. Otydligt tal försvårar kommunikationen med omgivningen, och de flesta med syndromet har tal-, språk- och kommunikationssvårigheter. Förmågan att förstå är större än den egna uttrycksförmågan, och personer med syndromet har ofta en god social förmåga. Trots koncentrationssvårigheter kan de ha ett gott minne, speciellt för personer och händelser.
Alla får näthinnesjukdomen retinitis pigmentosa, men synnedsättningen blir i allmänhet inte så uttalad.
Epilepsin och de strokeliknande anfallen brukar helt eller delvis gå tillbaka, medan de motoriska svårigheterna, som balans- och koordinationsproblem, håller sig på en stabil nivå. Nedsatt funktion i de perifera nerverna (neuropati) kan medföra muskelförtvining. Graden av skelettförändringar varierar från knappt märkbara till mycket påtagliga.
Sjukdomens påverkan på levern brukar minska med stigande ålder.
Andra former av PMM2-CDG
Det har blivit vanligare att sjukdomen har en annan symtombild än den klassiska. Oftast är det då en lindrigare variant som inte följer den vanliga stadieindelningen och inte heller ger samma grad av kognitiv funktionsnedsättning. Patienter diagnosticeras då i allmänhet senare i livet.
Diagnostik
Eftersom CDG-syndromen har en så varierad symtombild bör man försöka utesluta dessa syndrom hos alla med oklar neurodegenerativ sjukdom, framför allt när det samtidigt finns symtom från andra organ.
För att påvisa de flesta formerna (cirka 70) av CDG-syndromen kan man undersöka underglykosylering av transferrin. Tranferrin är ett serumprotein som hos friska personer bär två komplexa kolhydratkedjor. Vid CDG typ I (där PMM2-CDG ingår) är transferrin oftast underglykosylerat, vilket innebär att ett antal av transferrinmolekylerna har färre än två kedjor. Det finns ett flertal analyser för att undersöka graden av underglykosylering av transferrin, vanligast är så kallat CDT-test (carbohydrate deficient transferrin) som analyseras på ett flertal laboratorier. När detta test är positivt finns det indikation på att det kan röra sig om en CDG-sjukdom. Det är dock viktigt att utesluta andra sjukdomar (alkoholbruk hos ungdomar och vuxna, galaktosemi och hereditär fruktosintolerans) som också kan ge ett positivt CDT-resultat.
Om CDT-testet (eller andra underglykosyleringsmetoder) tyder på CDG-syndrom bör detta konfirmeras med analys av transferrin med hjälp av masspektrometri. Denna analysmetod är mer specifik och kan ge vägledning om vilket av CDG-syndromen det handlar om.
Om även masspektrometriprovet tyder på CDG typ I bör enzymanalys av PMM2 göras i antingen hudceller (fibroblaster) eller i vita blodkroppar. Enzymanalysen bekräftar om det är PMM2-CDG.
Mutationen i PMM2 kan därefter identifieras med hjälp av DNA-baserad diagnostik. I samband med att diagnosen ställs är det viktigt att familjen erbjuds genetisk vägledning. Genetisk vägledning omfattar information om sjukdomen och dess ärftlighetsgång, bedömning utifrån släktträdsanalys av sannolikheten för andra i familjen att insjukna samt information om diagnostik och behandling. Anlagsbärar- och fosterdiagnostik, liksom preimplantatorisk genetisk diagnostik (PGD) i samband med provrörsbefruktning, är möjlig om mutationen i familjen är känd.
Behandling/åtgärder
Det finns ännu ingen botande behandling för PMM2-CDG, utan insatserna inriktas på att förebygga de akuta medicinska situationer som sjukdomen ofta leder till samt habilitering för att stimulera barnets utveckling och kompensera för funktionsnedsättningarna. En tidig diagnos är mycket viktig för att få de bästa förutsättningarna att klara den första, ofta dramatiska, tiden.
Eftersom det behövs kontakt med många inom vården bör kontroll och behandling samordnas av en barnläkare med en helhetssyn på barnet. Barn med syndromet behöver hjälp av olika specialister, till exempel barnläkare inriktade på medicin, neurologi och ortopedi, samt stöd av ett habiliteringsteam.
Medicinsk uppföljning och behandling
PMM2-CDG är en sjukdom som kan drabba många vävnader och organ. Det är därför viktigt med regelbunden uppföljning och kontroll. Under de första åren behövs oftast behandling mot många olika symtom och komplikationer, som nedsatt leverfunktion, vätska i hjärtsäcken, strokeliknande anfall, epilepsi och koagulationsstörningar.
Om barnen har ätsvårigheter bör de få träffa en logoped för munmotorisk träning och en dietist för att få näringstillskott, men det är inga särskilda restriktioner vad gäller dieten för barn med syndromet. Vid stora problem med att maginnehållet tränger upp i matstrupen (reflux) och kräkningar kan det hjälpa med förtjockningsmedel eller syraneutraliserande medel (antacida). Att sitta upp efter att man har ätit kan också förhindra reflux. Är besvären svåra kan barnen tidvis behöva matas med en sond genom näsan eller via en ”knapp” (PEG, perkutan endoskopisk gastrotomi; operativt åstadkommen förbindelse mellan bukvägg och magsäck). Det är i så fall viktigt att fortsätta stimulera munmotoriken.
Nedsatt funktion av könskörtlarna kan med tiden medföra att skelettet urkalkas och hos vuxna med syndromet ge en ökad risk för frakturer. Med hjälp av hormoner kan pubertetsutvecklingen hos flickorna sättas igång. Vid upprepade frakturer kan behandling med kalktabletter och/eller bisfosfonat övervägas.
Hjärtat behöver kontrolleras varje år upp till fem års ålder för att vätska i hjärtsäcken eller påverkan på hjärtmuskeln ska kunna upptäckas. Vätska i hjärtsäcken kan behandlas med dränage och med läkemedel som acetylsalicylsyra eller kortison. En operation där hjärtsäcken tas bort (perikardiektomi) kan bli aktuell i svåra fall.
Leverns funktion behöver också kontrolleras regelbundet var sjätte månad (ALAT, albumin, PK). Ibland kan det behövas behandling med vitamin K och tillskott av proteinet albumin, som då ges direkt i en ven (intravenöst).
Störningar i blodets levringsförmåga (koagulationsrubbningar) medför en ökad risk för proppbildning eller blödning. Det är därför viktigt att kontrollera koagulationsvärdena och vara medveten om den ökade risken för blödningar i samband med operationer och tandbehandlingar. Inför ett kirurgiskt ingrepp ska blodets levringsförmåga undersökas för att ta reda på om det är nödvändigt med förebyggande behandling. I sådana fall behövs kontakt med ett koagulationscentrum).
Barnen bör också ha kontakt med en barntandvårdsspecialist (pedodontist) samt förstärkt förebyggande tandvård.
Det är viktigt med uppföljning varje år av ögonförändringar och synförmåga, även i vuxen ålder. Många med syndromet har brytningsfel och behöver glasögon. Skelning kan behandlas genom att barnet får bära en lapp över det öga som inte skelar, för att träna det öga som skelar. I svårare fall kan en operation mot skelning behövas. Dåligt mörkerseende till följd av förändringar i näthinnan kan kräva särskild belysning och för att undersöka näthinnans funktion bör barnen genomgå undersökning med elektroretinografi (ERG).
Strokeliknande episoder behandlas med rikligt med vätska intravenöst. Vid täta episoder kan det bli aktuellt med förebyggande (profylaktisk) behandling med acetylsalicylsyra i låga doser. Då måste koagulationsvärdena först kontrolleras.
Epilepsi behandlas med läkemedel.
Habilitering
Barn och ungdomar med PMM2-CDG behöver tidigt habiliteringsinsatser. I ett habiliteringsteam ingår yrkeskategorier med särskild kunskap om funktionsnedsättningar och deras effekter på vardagsliv, hälsa och utveckling. Insatserna sker inom det medicinska, pedagogiska, psykologiska, sociala och tekniska området. De består bland annat av utredning, behandling, utprovning av hjälpmedel, information om funktionsnedsättningen och samtalsstöd. Information om samhällets stöd samt råd inför anpassning av bostaden och andra miljöer som barnet vistas i ges också. Föräldrar, syskon och andra anhöriga kan också få stöd.
Alla insatser planeras utifrån varje barns och familjs behov och sker alltid i nära samverkan med personer i barnets nätverk.
Regelbunden kontakt med en fysioterapeut behövs för att stimulera och följa upp den motoriska utvecklingen och förebygga felställningar. Flera rapporter visar att de barn med CDG-syndrom som får aktiv sjukgymnastik klarar sig bättre. För att så långt som möjligt undvika sned rygg (kyfos och skolios) behövs oftast också korsett. Om felställningen är uttalad kan den behöva åtgärdas genom en ortopedisk operation. Skenor (ortoser) för underbenen kan underlätta gången och förhindra att musklerna förkortas och att rörligheten i lederna blir inskränkt (kontrakturer).
Den motoriska funktionsnedsättningen är vanligtvis stor, men med bra hjälpmedel och träning kan barnen utvecklas så att de klarar fler vardagliga saker, som toalettbesök, att tvätta och klä sig samt äta själva. Med rullstol, rollator eller andra gåhjälpmedel kan de öka sin rörlighet. Familjens bostad och bil behöver också anpassas för att underlätta vardagen.
Eftersom barn med PMM2-CDG kan ha varierande grad av utvecklingsstörning är det viktigt att vid upprepade tillfällen bedöma deras utvecklingsnivå.
Tal-, språk- och kommunikationsträning är viktig för barnens utveckling, och de behöver lära sig att kommunicera i olika sociala situationer. Kommunikationshjälpmedel och träning i att kommunicera med tecken som stöd eller att använda sig av alternativ och kompletterande kommunikation (AKK) kan behövas. AKK är ett samlingsnamn för kommunikation som inte bygger på tal. Särskilda pedagogiska insatser och tekniska hjälpmedel, till exempel datorer, behövs för att ta till vara barnens resurser.
För att underlätta vardagslivet kan kommunen erbjuda stöd i olika former. Exempel på sådant stöd är avlösning och personlig assistans. Familjen kan också ha behov av hjälp med samordningen av olika insatser.
De flesta vuxna med PMM2-CDG behöver fortsatt stöd från ett habiliteringsteam och i det dagliga livet, till exempel genom en bostad med särskild service eller personlig assistans samt daglig verksamhet.
Version 3.0