Sjukdom/skada/diagnos
MPI-CDG (tidigare CDG-Ib) är en sjukdom i en gruppen biokemiska ämnesomsättningssjukdomar som först beskrevs på 1980-talet, gemensamt kallade CDG-syndromen. CDG är en förkortning av det engelska namnet ’congenital disorders of glycosylation’, som betyder medfödda glykosyleringsstörningar.
För närvarande (2014) finns ett drygt 100-tal olika former av CDG-syndromen beskrivna. MPI-CDG ingår i en grupp av CDG-syndromen, kallad N-glykosyleringsstörningar, som idag omfattar cirka 50 olika former.
Vid MPI-CDG kan inte sockerarten fruktos-6-fosfat omlagras till sockerarten mannos-6-fosfat som används vid bildandet av komplexa sockerkedjor (oligosackarider). Detta leder till symtom från framför allt levern, bukspottkörteln och mag-tarmkanalen. Sjukdomen är den enda av CDG- syndromen för vilken det finns en verksam behandling (kosttillägg av sockret mannos). Efter puberteten är sjukdomen oftast helt stabil, men obehandlad kan den vara livshotande under barnaåren.
I Socialstyrelsens databas om ovanliga diagnoser finns även information om PMM2-CDG (länk) och ett översiktsdokument om CDG-syndromen (länk).
Förekomst
Det finns inga siffror för förekomsten, men sjukdomen har beskrivits hos ungefär 50 barn. I Sverige och Danmark har ett fåtal barn diagnostiserats och behandlats de senaste 10 åren.
Orsak till sjukdomen/skadan
De olika CDG-syndromen beror på fel i en process som kallas glykosylering. Glykosylering innebär att vissa proteiner och fetter (lipider) blir bärare av komplexa sockerarter (glykoproteiner respektive glykolipider). Glykoproteinerna består av en proteindel på vilken sockermolekyler som sticker ut som små antenner är fästade. Störningen i glykoliseringsprocessen gör att många viktiga glykoproteiner och glykolipider i kroppen blir felaktigt uppbyggda och delvis saknar sina sockerantenner.
MPI-CDG orsakas av en förändring (mutation) i genen MPI (15q24.1), som är en mall för tillverkningen av (kodar för) enzymet fosfomannosisomeras (PMI). Enzymer är proteiner som reglerar hastigheten av olika kemiska reaktioner i kroppen utan att själva förbrukas. PMI omvandlar sockermolekylen fruktos-6-fosfat till mannos-6-fosfat. Detta mannos-6-fosfat används sedan vid uppbyggnaden av komplexa sockerkedjor. Mannos-6-fosfat kan även bildas i kroppen från fritt mannos från födan eller från återanvändning av mannos i glykoproteiner. Under fosterlivet får fostret tillräckligt med mannos från modern för att kunna kompensera för bristen på PMI och får därför, till skillnad från de flest andra CDG-syndromen, vilket gör att hjärnans utveckling inte påverkas.
Typen av kolhydratdefekt skiljer sig åt mellan de olika formerna av N-glykosyleringsstörning. Numera delas sjukdomsformerna in i två grupper, typ I och typ II, beroende på om felet uppstått före eller efter att sockermolekylen bundits till proteinet. Typ I och II delas i sin tur in i olika undergrupper. MPI-CDG tillhör gruppen Typ I.
Ett femtontal olika mutationer är beskrivna i MPI, varav de flesta är punktmutationer som leder till ett PMI med försämrad funktion (missense mutations).
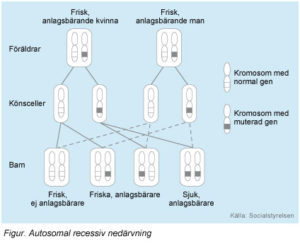
Ärftlighet
MPI-CDG och nästan alla andra CDG-syndrom nedärvs autosomalt recessivt, vilket innebär att båda föräldrarna är friska bärare av en förändrad (muterad) gen. Vid varje graviditet med samma föräldrar är sannolikheten 25 procent att barnet får den muterade genen i dubbel uppsättning (en från varje förälder). Barnet får då sjukdomen. Sannolikheten att barnet får den muterade genen i enkel uppsättning är 50 procent. Då blir barnet, liksom föräldrarna, frisk bärare av den muterade genen. Sannolikheten att barnet varken får sjukdomen eller blir bärare av den muterade genen är 25 procent.
I ovanliga fall har en av mutationerna uppstått i en könscell (germline mutation) och risken för återupprepning är då i princip ingen. Det är därför viktigt att även föräldrarna erbjuds genetisk utredning i samband med att MPI-CDG diagnosticeras (se under rubriken Diagnostik).
Symtom
Störningen i ämnesomsättningen leder till att inte tillräckligt många komplexa sockerkedjor kan bildas och ett antal glykoproteiner blir underglykosylerade. Vid MPI-CDG skyddas fostret av moderns mannos i blodet och har därför ingen underglykosylering under fosterlivet. Detta räddar bland annat hjärnan från skador under fosterutvecklingen (i motsats till de flesta CDG-syndromen).
Barn med MPI-CDG föds vanligtvis efter en normal graviditet och har inga symtom på sjukdom vid födseln. De flesta går igenom nyföddhetsperioden utan symtom och har en normal motorisk och kognitiv utveckling.
Symtomen som beskrivs nedan varierar mycket från person till person, och hos syskon med samma mutation kan sjukdomsbilden se mycket olika ut. Orsaken till detta är i nuläget oklar. Ofta utlöses symtomen i samband med stress på kroppens system som infektioner och tillväxt. Många är helt symtomfria i perioder.
Magtarmkanalen
De flesta barn med MPI-CDG har symtom från mag-tarmkanalen. Symtomen kan visa sig så tidigt som vid en månads ålder och bestå av:
Oförklarade upprepade episoder med kräkningar (cykliska kräkningar) och svår, ibland kronisk, diarré, vilket kan leda till upprepade sjukhusinläggningar med behov av nutrition via blodet (parenteral nutrition).
Ofta ses mycket låga nivåer av proteinet albumin i blodet och detta kan behöva kompenseras med upprepade infusioner. Det låga albuminvärdet leder ibland till svåra vätskeansamlingar i vävnaden (ödem) och kan ge problem med andningen och med att ta upp födoämnen ur kosten. Orsaken till det låga albuminvärdet kan vara leversjukdom som leder till en bristande produktion av albumin. Det är dock viktigt att utesluta ett annat tillstånd som kan leda till låga albuminvärden vid MPI-CDG, protein-förlorande enteropati (PLE). Vid PLE förloras proteiner (inklusive albumin) från blodet ut i tarmen, ett tillstånd som är synnerligen allvarligt och som obehandlat ofta leder till döden.
Levern
De flesta barn med MPI-CDG har tecken på leversjukdom med kroniskt lätt förhöjda leverprover (ALAT). Vid undersökning av en bit av levern i mikroskop (biopsi) syns oftast tecken på fibros, det vill säga ansamling av bindväv i levern som påverkar dess funktion. Hos några har levern omvandlats till skrumplever.
Störningar i blodets förmåga att koagulera är vanliga, och oftast är nivåerna av flera koagulationsfaktorer låga. Vid MPI-CDG ger detta oftast en risk för blodproppar (oftast i venerna), och en hos del barn med MPI-CDG har blodproppar varit det första tecknet på sjukdomen. Något enstaka barn har utvecklat stroke i samband med en propp i en artär i hjärnan.
Hormonella störningar
Många barn med MPI-CDG har tecken till för hög produktion av ämnet insulin som styr blodsockernivåerna. Blodsockret blir därför för lågt (hypoglykemi), framför allt i nyföddhetsperioden. Eftersom hypoglykemi också är vanligt hos friska nyfödda missas ofta diagnosen MPI-CDG.
Neurologiska symtom
Eftersom barn med MPI-CDG skyddas under fosterlivet av mannosnivåerna i moderns blod ger sjukdomen inte några symtom från centrala nervsystemet. Ett fåtal barn har dock utvecklat stroke efter proppar i hjärnans kärl (antingen artärer eller hjärnans sinussystem), vilket bland annat kan leda till halvsidig muskelsvaghet och problem med talet.
Vuxenlivet
Barn med MPI-CDG genomgår normal pubertet och efter detta stabiliseras sjukdomen. Oftast försvinner symtomen helt och behandlingen med mannos (se under Behandling) kan avslutas. Det finns ingen information som talar för att MPI-CDG skulle påverka fertiliteten, och det finns beskrivet att åtminstone en kvinna med sjukdomen har fått flera friska barn.
På senare år har ett antal personer med MPI-CDG upptäckts genom hälsokontroller (genom analys av CDT för att utesluta alkoholbruk), som trots tydliga tecken på underglykosylering helt har saknat symtom under barnaåren. Orsaken till att de inte har haft några symtom är i nuläget inte känd.
Diagnostik
MPI-CDG ska misstänkas hos alla med oklar proppsjukdom, kronisk leversjukdom eller svåra diarrétillstånd utan förklaring, framför allt om det finns tecken till proteinförlust via tarmen.
För att påvisa de flesta formerna (cirka 70) av CDG-syndromen kan man undersöka underglykosylering av transferrin. Tranferrin är ett serumprotein som hos friska personer bär två komplexa kolhydratkedjor. Vid CDG typ I (där MPI-CDG ingår) är transferrin oftast underglykosylerat, vilket innebär att ett antal av transferrinmolekylerna har färre än två kedjor. Det finns ett flertal analyser för att undersöka graden av underglykosylering av transferrin, vanligast är så kallat CDT-test (carbohydrate deficient transferrin) som analyseras på ett flertal laboratorier. När detta test är positivt finns det indikation på att det kan röra sig om en CDG-sjukdom. Det är dock viktigt att utesluta andra sjukdomar (alkoholbruk hos ungdomar och vuxna, galaktosemi och hereditär fruktosintolerans) som också kan ge ett positivt CDT-resultat.
Om CDT-testet (eller andra underglykosyleringsmetoder) tyder på CDG-syndrom bör detta konfirmeras med analys av transferrin med hjälp av masspektrometri. Denna analysmetod är mer specifik och kan ge vägledning om vilket av CDG-syndromen det handlar om.
Om även masspektrometriprovet tyder på CDG typ I bör enzymanalys av PMI göras i antingen hudceller (fibroblaster) eller i vita blodkroppar. Enzymanalysen bekräftar om det är MPI-CDG.
Mutationen i MPI kan därefter identifieras med hjälp av DNA-baserad diagnostik. I samband med att diagnosen ställs är det viktigt att familjen erbjuds genetisk vägledning. Genetisk vägledning omfattar information om sjukdomen och dess ärftlighetsgång, bedömning utifrån släktträdsanalys av sannolikheten för andra i familjen att insjukna samt information om diagnostik och behandling. Anlagsbärar- och fosterdiagnostik, liksom preimplantatorisk genetisk diagnostik (PGD) i samband med provrörsbefruktning, är möjlig om mutationen i familjen är känd.
Behandling/åtgärder
Tillskott av sockret mannos till födan har varit en effektiv behandling av de flesta symtomen vid MPI-CDG i de patientfall som rapporterats i litteraturen. Ofta förbättras besvären från mag-tarmkanalen efter några veckors behandling, och koagulationsfaktorerna återgår till normala nivåer efter någon månads behandling. En eventuell leverfibros kan dock ibland försämras trots behandlingen med tillskott av mannos. Eftersom ett flertal barn med MPI-CDG har avlidit på grund av sin sjukdom rekommenderas mannosbehandling till alla barn med sjukdomen.
Behandling med mannos bör ske via mag-tarmkanalen och inte via blodet (som en infusion) eftersom det finns beskrivningar av hur infusionsbehandling med mannos har lett till medvetslöshet och kramper, troligen orsakat av energibrist inne i cellerna (så kallad honey-bee effect). Tillståndet har kunnat hävas med infusion av höga mängder av sockret glukos.
Vid svår PLE kan behandling med läkemedlet heparin också behövas och bör inledas vid en specialistklinik.
Medicinsk uppföljning och behandling
Leverns funktion behöver kontrolleras regelbundet varje år (ALAT, albumin, PK). Ibland kan det behövas behandling med vitamin K och tillskott av proteinet albumin, som då ges direkt i en ven (intravenöst). Kontrollerna behöver ske oftare om det finns tecken till PLE eller kroniska diarrébesvär.
Vid skrumplever kan det bli aktuellt med levertransplantation.
Störningar i blodets levringsförmåga (koagulationsrubbningar) medför oftast en ökad risk för proppbildning för personer med MPI-CDG. Det är därför viktigt att koagulationsvärdena kontrolleras och att det finns en medvetenhet om den ökade risken för proppar och/eller blödningar i samband med operationer. Även vid tandbehandlingar kan det finnas risk för blödningar. Inför ett kirurgiskt ingrepp ska blodets levringsförmåga undersökas för att ta reda på om det är nödvändigt med förebyggande behandling.
Version 3.0